Pulmonary arterial hypertension (PAH) is a progressive disease that reduces blood flow and increases pressure in the arteries of the lungs.
What causes PAH?
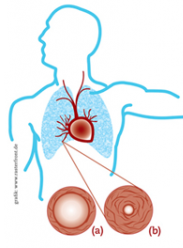
The pulmonary arteries are the large blood vessels responsible for transporting blood from the heart to the lungs to pick up oxygen. In PAH, the cells lining the inside of these arteries enlarge and multiply. As a result, the walls of the arteries may constrict and thicken, causing resistance to blood flowing through them and so increasing blood pressure. The right side of the heart has to work harder to pump blood through the arteries. This places an increasing strain on the heart, which causes it to enlarge and can lead to right heart failure.1
LifeART Collection Images Copyright © 1989-2001 Lippincott Williams & Baltimore, MD
(a) Cross section of a pulmonary artery in a healthy person: the vessel walls are flexible and elastic, allowing blood to pass through easily.
(b) Cross section of a pulmonary artery in a person with PAH: thickening of the walls of vessel creates resistance to blood flow, making it difficult for the heart to pump blood through the lungs.
How many people are affected by PAH?
PAH affects 15–50 individuals per million people.2 People of all ages and ethnic backgrounds suffer from this disease, although it is more common in women than men.3
PAH is defined as ‘Group 1’ in the World Health Organization (WHO) clinical classification of pulmonary hypertension (PH)4 (see classification for further information).
The future for PAH
Although a cure for this life-threatening disease is still some way off5, there is much to be optimistic about. An ever-increasing number of treatments are becoming available that improve both quality and length of life for patients with PAH. The present aim is to ensure that all patients with PAH have access to centers of excellence in the diagnosis, management and ongoing treatment of their disease. In case of inherited disease research on gene therapy has started a while ago.
Reviewed by Prof. Simon J Gibbs
Last medical update: 05/30/2020
Sources
- Rosenblum WD. Pulmonary arterial hypertension: pathobiology, diagnosis, treatment, and emerging therapies. Cardiol Rev 2010;18:58–63.
- Peacock AJ, Murphy NF, McMurray JJ et al. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007;30:104–9.
- “Pulmonary arterial hypertension“. Genetics Home Reference. January 2016. Archived from the original on 28 July 2017. Retrieved 30 July 2017.
- Simonneau G, Gatzoulis MA, Adatia I et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62:D34–41.
- “What Is Pulmonary Hypertension? – NHLBI, NIH“. NHLBI. 2 August 2011. Archived from the original on 28 July 2017. Retrieved 30 July 2017.
- Reynolds, PN (February 2011). “Gene therapy for pulmonary hypertension: prospects and challenges”. Expert Opinion on Biological Therapy. 11 (2): 133–43. doi: 10.1517/14712598.2011.542139. PMID 21219232.
Further information and educational materials on PAH can be found in the section ‘PH. It’s personal’.